Anisocoria: difference in the pupillary size
History taking:
Ask who first noticed the anisocoria and attempt to establish the duration. Review of old photos. Magnification may be required to assess pupil size. Exclude a history of neck or chest surgery/ injury, limb weakness, difficulty focusing, diplopia, and ptosis.
Dr.fawaz al sarayreh lecture
Examination:
Identify which pupil is abnormal by examining first in the dark then in the light. Greater anisocoria in the dark indicates impaired dilatation (sympathetic dysfunction): greater anisocoria in the light indicates impaired constriction (parasympathetic dysfunction). Physiological anisocoria is usually ≤2 mm and the difference is the same at all light levels. Examine the pupils and the iris on the slit lamp.
Differential diagnosis of abnormally large pupil:
- Adie’s pupil (below)
- 3rd nerve palsy,
- dilating drops,
- traumatic mydriasis,
- iris rubeosis,
- Urrets-Zavalia syndrome (iris atrophy following corneal graft),
- physiological anisocoria.
■ Adie’s tonic pupil (Holmes-Adie pupil): Presumed postviral denervation of the sphincter pupillae and ciliary muscle produces anisocoria and difficulty focusing. Owing to reinnervation, accommodation recovers in a few weeks but the sphincter pupillae becomes partially innervated by lens fibres.
Signs thereafter include light-near dissociation (slow or absent constriction to light but prompt constriction on attempted near vision), segmental sphincter palsy on slit lamp examination which gives rise to so-called vermiform movements of the iris, and absent limb reflexes. Slow dilation following accommodation (tonic constriction) differentiates it from Argyll Robertson pupils. Exclude ptosis and diplopia (3rd nerve palsy). Whereas normal pupils do not usually constrict to G. pilocarpine 0.125%, Adie’s pupil does (denervation hypersensitivity), but this test has only moderate specificity, as preganglionic lesions do likewise. Further investigations are not normally required. Arrange routine referral to a neuro-ophthalmologist. Long-term follow-up is not required. It is often bilateral but asymmetric. The affected pupils eventually
become small.
Differential diagnosis of an abnormally small pupil Consider Horner’s syndrome (below), pilocarpine drops, uveitis/posterior synechiae, chronic unilateral aphakia, and
physiological anisocoria.
■ Horner’s syndrome: Sympathetic denervation produces miosis and mild ptosis. It may be preganglionic or postganglionic.
Preganglionic (central) causes include lung and breast malignancy, sympathetic chainschwannoma, and cervical spine damage (e.g. C8 or T1 disc prolapse).
Postganglionic causes include internal carotid dissection, neck tumours, cavernous sinus disease (especially if 6th nerve palsy coexists), and cluster headache.
The diagnosis of Horner’s syndrome is Confirmed by Continued pupil Constriction despite G. Cocaine 4% (delay further pharmacological testing for 2 days after cocaine testing). Preganglionic causes produce ipsilateral anhydrosis of the face (ptosis, miosis, anhydrosis) and the pupil dilates with G. hydroxyamphetamine 1% (no effect if postganglionic).
Investigations: depend on the likely cause, but if painful request urgent T2 weighted MRI axial scans to exclude carotid dissection. This is associated with a high risk of embolic stroke within 10 days and anticoagulation is indicated.
Congenital preganglionic Horner’s syndrome typically has iris heterochromia and is seen on old photographs. Acquired childhood cases require investigation to exclude neoplasia, particularlarly cervicothoracic neuroblastoma, though most are benign.
Transient Visual Loss
History Ask about: duration; whether one or both eyes are affected; total blackness (arterial occlusion) or just blurred; patchy grey blobs (spasm of choroidal vessels); cardiovascular risk factors; TIAs or strokes; known carotid disease; headache; migrainous aura; dizziness, hearing, or speech problems; loss of balance; haloes; eye pain; abnormal clotting (DVTs); scalp tenderness; jaw claudication (over 50 years).
Specific precipitants may suggest the diagnosis:
■ Bright light: chronic retinal ischaemia due to carotid insufficiency.
■ Eye movements: space-occupying orbital lesions or an optic nerve tumour.
■ Prolonged reading or evening onset: intermittent angle-closure glaucoma.
■ Exercise: pigment dispersion syndrome.
■ Standing up: usually indicates reduced perfusion pressure including postural hypotension, carotid insufficiency, and giant cell arteritis (precedes nerve infarction). Papilloedema may produce brief monocular or bilateral obscurations with either standing up or stooping down.
Examination:
Check BP in both arms (sitting and standing if appropriate); radial pulse; cardiac and carotid auscultation; temporal artery palpation (in patients over 50 years); VA; confrontation visual fields; colour vision; RAPD; corneal clarity (oedema or endothelial pigment); iris rubeosis; gonioscopy (is angle closeable?); IOP; dilated fundoscopy (especially retinal vessels for emboli, venous dilation, retinal haemorrhages and optic disc for swelling); assess central retinal artery perfusion pressure; other tests as indicated.
Differential diagnosis
■ The following are the more common or serious causes:
1. Carotid or cardiac emboli
2. Carotid dissection: may have neck pain and Horner’s syndrome.
3. Migraine: usually hemianopia, positive features, with zigzags typical.
4. Giant cell arteritis
5. Vertebrobasilar ischaemia
6. Intermittent angle-closure glaucoma
7. Occipital embolus: may cause hemianopia loss or transient blindness.
8. Retinal arterial embolus: typically, a curtain descent to a blackout.
9. Retinal vasospasm: more likely a whiteout.
10. Choroidal vasospasm: vision disappears in patches.
11. Cardiac dysrhythmia: blindness may precede loss of consciousness or occur in isolation.
12. Occipital epilepsy: usually hemianopia with positive symptoms such as coloured circles.
Management Investigation and treatment depends on the likely cause. In the case of transient monocular blindness, emboli usually arise from the aorta, carotids, or heart valves. Most commonly, they are cholesterol, platelet-fibrin, or calcific, but septic, amniotic fluid, air, fat or talc (i.v. drug users) may rarely occur in specific situations. If likely, start oral aspirin 75 mg o.d.
Visual Field Defects
The accurate delineation of visual field defects is critical to the diagnosis of visual pathway lesions. Visual field defects are frequently asymptomatic and may be detected on routine screening (usually by an optometrist) or when field tests are preformed for some other reason. An awareness of the various artefactually produced field defects is important.
Symptoms Patients are less likely to notice field defects from optic nerve or visual pathway lesions if these spare the central field. Retinal lesions often produce positive scotomas with patients aware of photopsia within the visual field defect.
History: ِِAsk when and how the field defect was first noticed. Sudden onset or gradual? Any recovery? Ask about cardiovascular risk factors, photopsia, pain, headache and other neurological symptoms, and symptoms of pituitary disease (amenorrhoea, hypothyroidism, loss of libido, headache, and acromegaly).
Examination:
Check BP, cranial nerves, VA, colour vision, RAPD, formal fi elds, eye movements, IOP, assess angle, and dilated fundoscopy. Exclude ptosis, and disc cupping, pallor, or swelling. Many field defects are relative, and not absolute.
Differential diagnosis Abnormal visual fields may be caused by retinal pathology (e.g. retinal detachment or vein occlusion).
A cataract may cause a globally decreased field but not focal defects. A homonymous hemianopia should not cause a decreased VA. Glaucoma can cause a range of field defects but confirm that the field defect corresponds to the sectoral neuroretinal rim thinning; colour vision is relatively well preserved until late in the disease, unlike optic nerve disease.
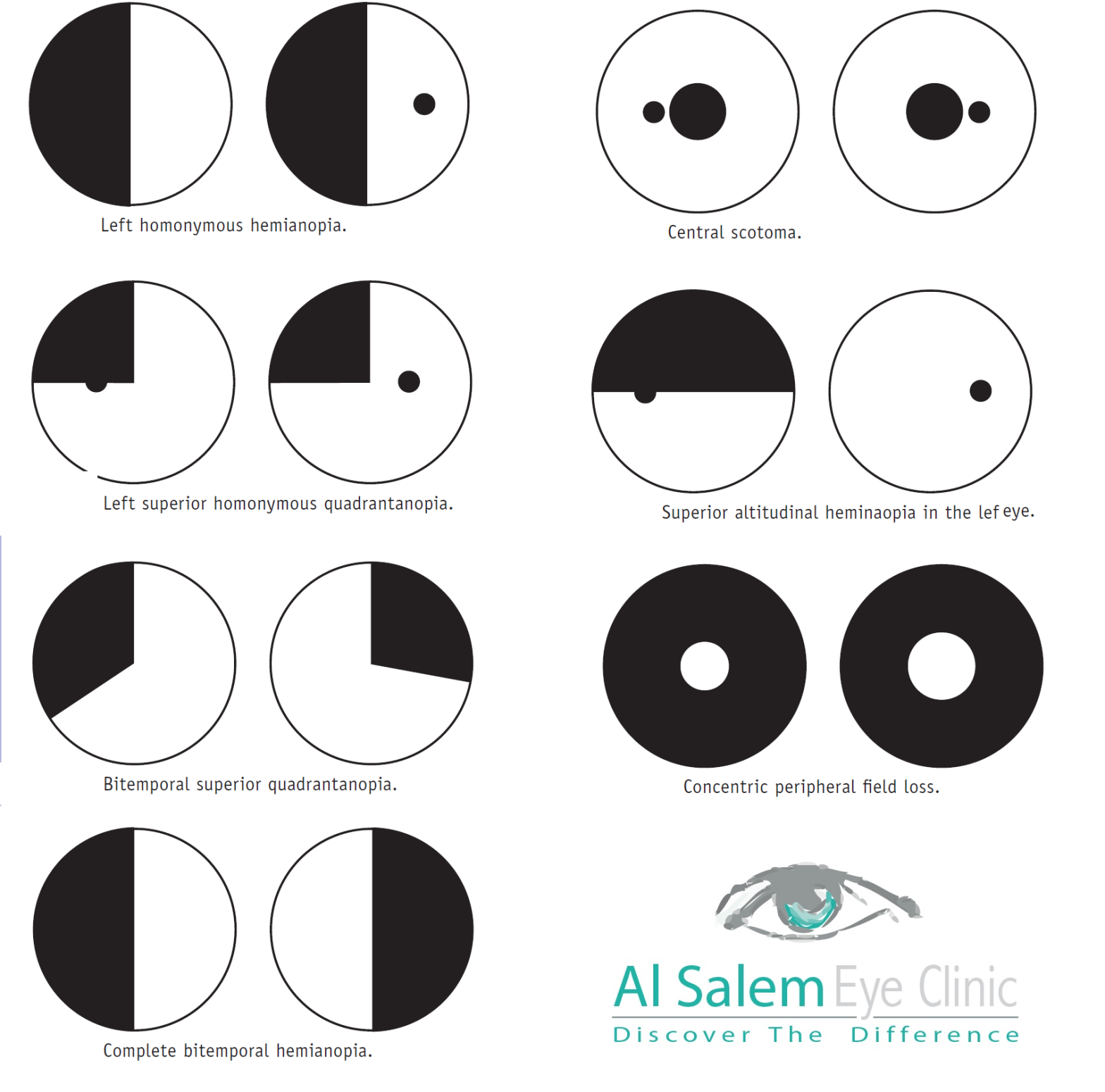
■ Left homonymous hemianopia : consider a right postchiasmal lesion such as occipital lobe CVA or tumour. The more congruous the field defect, the nearer to the occipital lobe, but a large lesion affecting both temporal and parietal lobes (the entire optic radiation) could also cause this.
■ Left superior homonymous quadrantanopia: probably right inferior occipital cortex but consider right temporal lobe lesion. Inferior homonymous quadrantanopia may be caused by a parietal lobe lesion. The defect may be relative or absolute. The vertical meridian will be absolutely respected but usually not the horizontal meridian.
■ Bitemporal superior quadrantanopia: typically caused by pituitary tumours but will be relative; the defect will respect the vertical meridian but not the horizontal meridian.
As the defect progresses, the bitemporal hemianopia will become more complete but usually asymmetric and eventually with evidence of optic neuropathy on one or other side. A craniopharyngioma may cause a bilateral inferotemporal quadrantanopia but is more likely to give rise to a combination of the optic nerve, chiasm and tract deficits.
■Complete bitemporal hemianopia: may occur with compressive lesions of the chiasm but such a clear-cut deficit is typically only seen in cases of traumatic chiasmal
transection.
■ Central scotoma: the commonest cause is age-related macular degeneration. In the case of optic nerve disease, such a symmetrical picture is more likely to be toxic, nutritional, or an inherited condition. Cone dystrophy can produce a similar picture.
■ Superior altitudinal heminaopia in the left eye: typically seen with nonarteritic anterior ischaemic optic neuropathy but also normal pressure glaucoma (usually
bilateral and with arcuate defects in the lower field also), hemicentral vein occlusion, branch retinal artery occlusion, ptosis (less severe), sector panretinal photocoagulation (PRP) laser, and inferior retinal detachment.
■ Concentric peripheral field loss: may be seen in retinitis pigmentosa, chronic atrophic papilloedema (e.g.idiopathic intracranial hypertension), end-stage glaucoma, PRP, central retinal artery occlusion with cilioretinal artery sparing (usually unilateral), optic neuropathies, and in vigabatrin toxicity.

